What is CrossMap ?
CrossMap is a program for genome coordinates conversion between different assemblies (such as hg18 (NCBI36) <=> hg19 (GRCh37)). It supports commonly used file formats including BAM, CRAM, SAM, Wiggle, BigWig, BED, GFF, GTF, MAF VCF, and gVCF.
How CrossMap works?

Release History
10/30/2025: Version 0.7.4
Fixed issue where removing the only ALT allele left the “ALT” field empty. - In this case, the variant will now be written to the unmapped variant file.
07/17/2024: Version 0.7.3
Fixed bugs in VCF and gVCF liftover. - For variants with multiple ALT alleles, remove the ALT allele that is identical to the REF allele.
05/09/2024: Version 0.7.2
Fixed bugs in VCF and gVCF liftover. - When insertion/deletion variants are mapped to the reverse strand of the target assembly, their REF alleles are now correctly updated.
# Input VCF (header not shown)
# These are hg19/GRCh37-based variants. They map to the reverse region on hg38/GRCh38.
chr7 61879851 rs1223781306 A AC . . .
chr1 145382743 rs782203468 G GA . . .
chr1 144852392 indel.6062 AC A . . .
chr1 145698920 indel.6189 TGCTTGGGGTGCTTACG T . . .
# Output (Note that REF alleles differ from input)
chr7 62217710 rs1223781306 T TG . . .
chr1 146052257 rs782203468 C CT . . .
chr1 149032049 indel.6062 GG G . . .
chr1 145736150 indel.6189 CCGTAAGCACCCCAAGC C . . .
01/11/2024: Version 0.7.0
Fixed bugs in VCF variant liftover.
Added handling for non-DNA ALT alleles (e.g.,
<DEL>).Replaced
setup.pywith pyproject.toml.
Note
Starting from v0.7.0, the main program CrossMap.py has been renamed to CrossMap
due to restrictions on using “.” in script names under pyproject.toml.
To maintain backward compatibility, add the following line to your ~/.bashrc file:
alias CrossMap.py='CrossMap'
07/21/2023: Version 0.6.4
Fixed bug when the sequence in a BAM file is represented as “*”.
Updated code style.
07/12/2022: Version 0.6.4
Fixed bug where input BigWig files lacked coverage signals for some chromosomes.
If a VCF file has no CONTIG field, the long chromosome ID (e.g., “chr1”) is now used by default.
03/04/2022: Version 0.6.3
Fixed bug in v0.6.2 where the alternative allele was empty.
02/22/2022: Version 0.6.2
For insertions and deletions, the first nucleotide of the ALT allele (the 5th VCF field) is now updated to the nucleotide at the POS of the reference genome.
11/29/2021: Version 0.6.1
Same as v0.6.0, but removes unused modules from the
libfolder.
11/16/2021: Version 0.6.0
Uses
os.path.getmtimeinstead ofos.path.getctimefor checking FASTA file timestamps.Added
--unmap-fileoption to theCrossMap.py bedcommand.
04/16/2021: Version 0.5.3 / 0.5.4
Added
CrossMap.py viewchaincommand to convert a chain file into a more readable, block-to-block format.
12/08/2020: Version 0.5.2
Added
--no-comp-allelesflag toCrossMap.py vcfandCrossMap.py gvcf. If set, CrossMap will not check whether the reference and alternative alleles differ.
08/19/2020: Version 0.5.1
In
CrossMap.py region, additional columns (beyond the 3rd) in BED files are now preserved after conversion.
08/14/2020: Version 0.5.0
Added
CrossMap.py regionfunction to convert large genomic regions as a whole. UnlikeCrossMap.py bed, which splits large regions, this function processes them intact.
07/09/2020: Version 0.4.3
Structural Variant VCF files often use the INFO/END field to indicate deletion endpoints. Version 0.4.3 updates the “END” coordinate accordingly.
05/04/2020: Version 0.4.2
Added support for GVCF file conversion.
03/24/2020: Version 0.4.1
Fixed issue with consecutive TAB characters in input MAF files.
10/09/2019: Version 0.3.8
UCSC chain files are now permanently removed, per request from UCSC, as they retain University of California copyright.
07/22/2019: Version 0.3.6
Added support for MAF (Mutation Annotation Format).
Fixed
TypeError: AlignmentHeader does not support item assignment (use header.to_dict())error in BAM liftover. - Users no longer need to downgradepysamto 0.13.0.
04/01/2019: Version 0.3.4
Fixed bugs where chromosome IDs in chain files lacked a “chr” prefix.
Added detection for inverted mappings and automatic reverse-complement of ALT alleles (thanks to Andrew Yates).
Improved wording.
01/07/2019: Version 0.3.3
Identical to Version 0.3.2. Released because
CrossMap-0.3.2.tar.gzwas corrupted on PyPI.
12/14/2018: Version 0.3.2
Fixed KeyError (e.g.,
KeyError: "sequence 'b'7_KI270803v1_alt'' not present"). - This occurred when a locus mapped to an “alternative,” “unplaced,” or “unlocalized” contig absent intarget_ref.fa. - Such loci are now skipped and saved to the “.unmap” file.
11/05/2018: Version 0.3.0
Added Python 3 support (v0.3.0 and later). - Previous versions supported only Python 2.7.*
Added dependency: pyBigWig.
Installation
Install from PyPI
To install the latest stable release from PyPI:
pip install CrossMap
Install from GitHub
To install the latest development version directly from GitHub:
pip install git+https://github.com/liguowang/CrossMap.git
Upgrade
To upgrade an existing installation to the newest version:
pip install --upgrade CrossMap
Uninstall
pip uninstall CrossMap
Input and Output
Chain File
A chain file describes a pairwise alignment between two reference genome assemblies.
These files are required by CrossMap to map genomic coordinates between assemblies.
Chain files are available from both UCSC and Ensembl.
UCSC Chain Files
From hs1 (T2T-CHM13) to hg38, hg19, mm10, or mm9 (and vice versa): https://hgdownload.soe.ucsc.edu/goldenPath/hs1/liftOver/
From hg38 (GRCh38) to hg19 and other organisms: http://hgdownload.soe.ucsc.edu/goldenPath/hg38/liftOver/
From hg19 (GRCh37) to hg17, hg18, hg38, and other organisms: http://hgdownload.soe.ucsc.edu/goldenPath/hg19/liftOver/
From mm10 (GRCm38) to mm9 and other organisms: http://hgdownload.soe.ucsc.edu/goldenPath/mm10/liftOver/
Ensembl Chain Files
Human to Human: ftp://ftp.ensembl.org/pub/assembly_mapping/homo_sapiens/
Mouse to Mouse: ftp://ftp.ensembl.org/pub/assembly_mapping/mus_musculus/
Other organisms: ftp://ftp.ensembl.org/pub/assembly_mapping/
User Input File
CrossMap supports the following input file formats:
Output File
The output file format depends on the input file type:
Input Format |
Output Format |
|---|---|
BED |
BED (genome coordinates will be updated) |
BAM |
BAM (genome coordinates, header section, all SAM flags, and insert sizes will be updated) |
CRAM |
BAM (requires |
SAM |
SAM (genome coordinates, header section, all SAM flags, and insert sizes will be updated) |
Wiggle |
BigWig |
BigWig |
BigWig |
GFF |
GFF (genome coordinates will be updated to the target assembly) |
GTF |
GTF (genome coordinates will be updated to the target assembly) |
VCF |
VCF (header section, genome coordinates, and reference alleles will be updated) |
GVCF |
GVCF (header section, genome coordinates, and reference alleles will be updated) |
MAF |
MAF (genome coordinates and reference alleles will be updated) |
Usage Information
To display the help message, run:
CrossMap -h
Example output:
usage: CrossMap [-h] [-v]
{bed,bam,gff,wig,bigwig,vcf,gvcf,maf,region,viewchain} ...
CrossMap (v0.7.3) is a program for converting (lifting over) genome coordinates
between different reference assemblies (e.g., from human GRCh37/hg19 to
GRCh38/hg38, or vice versa).
Supported file formats: BAM, BED, BigWig, CRAM, GFF, GTF, GVCF, MAF (mutation
annotation format), SAM, Wiggle, and VCF.
Positional arguments:
{bed,bam,gff,wig,bigwig,vcf,gvcf,maf,region,viewchain}
sub-command help
bed Convert BED, bedGraph, or other BED-like files.
Only genome coordinates (the first 3 columns) will be updated.
Regions mapped to multiple locations in the new assembly will be split.
Use the "region" command for large genomic regions, or "wig"
for bedGraph/BigWig output.
bam Convert BAM, CRAM, or SAM format files.
Genome coordinates, header section, all SAM flags, and insert sizes
will be updated.
gff Convert GFF or GTF format files.
Genome coordinates will be updated.
wig Convert Wiggle or bedGraph format files.
Genome coordinates will be updated.
bigwig Convert BigWig files.
Genome coordinates will be updated.
vcf Convert VCF files.
Genome coordinates, header section, and reference alleles will be updated.
gvcf Convert GVCF files.
Genome coordinates, header section, and reference alleles will be updated.
maf Convert MAF (Mutation Annotation Format) files.
Genome coordinates and reference alleles will be updated.
region Convert large genomic regions (in BED format), such as CNV blocks.
Genome coordinates will be updated.
viewchain Display the content of a chain file in a human-readable,
block-to-block format.
Convert BED Format Files
A BED (Browser Extensible Data) file is a tab-delimited text file describing genomic regions or gene annotations. Each line represents one feature and may contain between 3 and 12 columns.
CrossMap converts BED files between reference assemblies by updating chromosome and coordinate information while keeping other fields intact.
For BED files with fewer than 12 columns, only the chromosome and coordinate fields are updated.
For 12-column BED files, all columns are updated except: - 4th column: feature name - 5th column: score - 9th column: RGB display color
If any exon block in a 12-column BED file fails to map to the target assembly, the entire line will be skipped. Regions from the source assembly mapping to multiple positions in the target assembly are split into separate entries.
Input and Output
The input BED file can be one of the following:
Plain text file
Compressed file with extensions:
.gz,.Z,.z,.bz,.bz2, or.bzip2URL pointing to an accessible remote file (
http://,https://, orftp://)
Note: compressed remote files are not supported.
The output is a BED-format file with the same number of columns as the input.
BED Variants Supported
Standard BED format has 12 columns, but CrossMap also supports BED-like variants:
BED3: first three columns —
chrom,start,endBED6: first six columns —
chrom,start,end,name,score,strandOther BED-like formats: between 3 and 12 columns, where extra columns are arbitrary
Note
For BED-like formats, CrossMap updates only the
chrom,start,end, andstrandfields. All other columns are preserved as-is.Lines beginning with
#,browser, ortrackare ignored.Lines containing fewer than three columns are skipped.
The 2nd and 3rd columns must be integers.
The
+strand is assumed if no strand information is provided.For standard 12-column BED files, if any exon block cannot be uniquely mapped, the entire entry is skipped.
Both
input_chain_fileandinput_bed_filemay be regular or compressed files (.gz,.Z,.z,.bz,.bz2,.bzip2) and may also be local or remote URLs.If
output_fileis not specified, CrossMap prints results to the console. In this case, original (unconverted) entries are also printed.If an input region cannot be mapped contiguously to the target assembly, it is split.
The
*.unmapfile contains entries that could not be unambiguously converted.
Command Help
To view command-line help for BED conversion, run CrossMap bed -h
usage: CrossMap bed [-h] [--chromid {a,s,l}] [--unmap-file UNMAP_FILE]
input.chain input.bed [out_bed]
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
The chain file may be plain text or compressed
(.gz, .Z, .z, .bz, .bz2, .bzip2).
input.bed Input BED file. The first 3 columns must be
“chrom”, “start”, and “end”.
Supports plain text, compressed (.gz, .bz2, etc.),
or URL sources (http://, https://, ftp://).
Compressed remote files are not supported.
out_bed Output BED file.
If omitted, CrossMap writes to STDOUT.
options:
-h, --help Show this help message and exit
--chromid {a,s,l} Style of chromosome IDs:
a = as-is
l = long style (e.g., "chr1", "chrX")
s = short style (e.g., "1", "X")
--unmap-file UNMAP_FILE
File to save unmapped entries.
Ignored if [out_bed] is not provided.
Example 1 — Run Without Output File
Running CrossMap bed without specifying an output file prints the results to the screen.
$ cat Test1.hg19.bed
chr1 65886334 66103176
chr3 112251353 112280810
chr5 54408798 54469005
chr7 107204401 107218968
$ CrossMap bed GRCh37_to_GRCh38.chain.gz Test1.hg19.bed
2024-01-12 08:36:35 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
chr1 65886334 66103176 -> chr1 65420651 65637493
chr3 112251353 112280810 -> chr3 112532506 112561963
chr5 54408798 54469005 -> chr5 55112970 55173177
chr7 107204401 107218968 -> chr7 107563956 107578523
Note
The first three columns represent hg19-based coordinates, while the last three columns are hg38-based coordinates.
Example 2 — Save Output to File
When an output file is specified, CrossMap saves the converted results to that file.
$ CrossMap bed GRCh37_to_GRCh38.chain.gz Test1.hg19.bed output.hg38
2024-01-12 08:35:56 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
$ cat output.hg38
chr1 65420651 65637493
chr3 112532506 112561963
chr5 55112970 55173177
chr7 107563956 107578523
Note
Genomic intervals that fail to map will be saved in a separate file named output.hg38.unmap.
Example 3 — Split Non-Contiguous Mappings
If an input region cannot be mapped contiguously to the target assembly, it is automatically split.
$ cat Test2.hg19.bed
chr20 21106623 21227258
chr22 30792929 30821291
$ CrossMap bed GRCh37_to_GRCh38.chain.gz Test2.hg19.bed
2024-01-12 08:53:10 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
chr20 21106623 21227258 (split.1:chr20:21106623:21144549:+) chr20 21125982 21163908
chr20 21106623 21227258 (split.2:chr20:21144549:21176014:+) chr20 21163909 21195374
chr20 21106623 21227258 (split.3:chr20:21176014:21186161:+) chr20 21195375 21205522
chr20 21106623 21227258 (split.4:chr20:21186161:21227258:+) chr20 21205523 21246620
chr22 30792929 30821291 (split.1:chr22:30792929:30819612:+) chr22 30396940 30423623
chr22 30792929 30821291 (split.2:chr22:30819615:30821291:+) chr22 30423627 30425303
Note
As before, the first three columns are hg19-based, and the last three are hg38-based.
Example 4 — Converting bedGraph Files
A bedGraph file can be converted using either
CrossMap bed or CrossMap wig. The choice of command determines the output format:
Using
CrossMap bed→ output is a bedGraph file.Using
CrossMap wig→ output is a BigWig file.
$ head -3 Test3.hg19.bgr
chrX 2705083 2705158 1.0
chrX 2813094 2813169 0.9
chrX 2813169 2814363 0.1
...
$ CrossMap bed hg19ToHg38.over.chain.gz Test3.hg19.bgr
2024-01-12 09:05:05 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
chrX 2705083 2705158 1.0 -> chrX 2787042 2787117 1.0
chrX 2813094 2813169 0.9 -> chrX 2895053 2895128 0.9
chrX 2813169 2814363 0.1 -> chrX 2895128 2896322 0.1
...
$ CrossMap wig GRCh37_to_GRCh38.chain.gz Test3.hg19.bgr output.hg38
2024-01-12 09:09:52 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
2024-01-12 09:09:52 [INFO] Liftover wiggle file "Test3.hg19.bgr" to bedGraph file "output.hg38.bgr"
2024-01-12 09:09:52 [INFO] Merging overlapped entries in bedGraph file
2024-01-12 09:09:52 [INFO] Sorting bedGraph file: output.hg38.bgr
2024-01-12 09:09:52 [INFO] Writing header to "output.hg38.bw" ...
2024-01-12 09:09:52 [INFO] Writing entries to "output.hg38.bw" ...
Example 5 — Converting Large Genomic Regions
Use CrossMap region to convert large genomic regions (e.g.,
CNV blocks) from one assembly to another.
$ cat Test4.hg19.bed
chr2 239716679 243199373 # A large genomic interval of ~3.48 Mb
If you use the CrossMap bed command, this interval will be split multiple times:
$ CrossMap bed GRCh37_to_GRCh38.chain.gz Test4.hg19.bed
2024-01-12 09:12:45 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
chr2 239716679 243199373 (split.1:chr2:239716679:239801978:+) chr2 238808038 238893337
chr2 239716679 243199373 (split.2:chr2:239831978:240205681:+) chr2 238910282 239283985
chr2 239716679 243199373 (split.3:chr2:240205681:240319336:+) chr2 239283986 239397641
... (split 74 times)
To lift over the region as a whole, use the CrossMap region command.
By default, r = 0.85.
$ CrossMap region GRCh37_to_GRCh38.chain.gz Test4.hg19.bed
2024-01-12 09:15:24 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
chr2 239716679 243199373 -> chr2 238808038 242183529 map_ratio=0.9622
If we increase the required mapping ratio to r = 0.99, the region fails to lift over:
$ CrossMap region GRCh37_to_GRCh38.chain.gz Test4.hg19.bed -r 0.99
2024-01-12 09:18:53 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
chr2 239716679 243199373 Fail map_ratio=0.9622
Convert BAM / CRAM / SAM Format Files
The SAM (Sequence Alignment/Map) format is a widely used text-based format for storing sequence alignments. BAM is its binary, compressed version (Li et al., 2009), while CRAM provides a more efficient, reference-based alternative.
Most high-throughput sequencing (HTS) alignments are stored in SAM/BAM/CRAM format, and many downstream bioinformatics tools rely on these formats. CrossMap performs liftover of alignment files by updating:
Chromosome names
Genome coordinates
SAM header section (including version, chain file, and original file name)
All SAM flags and pairing information
For paired-end reads, insert sizes are automatically recalculated. The input BAM file must be sorted and indexed (e.g., using Samtools).
The output format is determined by the input format:
- SAM → SAM
- BAM → BAM (sorted and indexed automatically)
- CRAM → BAM (requires pysam >= 0.8.2)
Command Help
To view the help message for BAM/CRAM/SAM conversion, run CrossMap bam -h
usage: CrossMap bam [-h] [-m INSERT_SIZE] [-s INSERT_SIZE_STDEV]
[-t INSERT_SIZE_FOLD] [-a] [--chromid {a,s,l}]
input.chain input.bam [out_bam]
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
May be plain text or compressed (.gz, .Z, .z, .bz, .bz2, .bzip2).
input.bam Input BAM file (https://genome.ucsc.edu/FAQ/FAQformat.html#format5.1).
out_bam Output BAM file. If omitted, CrossMap writes to STDOUT.
options:
-h, --help Show this help message and exit
-m, --mean INSERT_SIZE
Average insert size for paired-end sequencing (bp).
-s, --stdev INSERT_SIZE_STDEV
Standard deviation of insert size.
-t, --times INSERT_SIZE_FOLD
A pair is considered "properly paired" if both reads
map to opposite strands and the distance between them
is less than ‘-t × stdev’ from the mean.
-a, --append-tags Add mapping status tags to each alignment.
See tag definitions below.
--chromid {a,s,l} Style of chromosome IDs:
a = as-is
l = long (e.g., "chr1", "chrX")
s = short (e.g., "1", "X")
Example
$ CrossMap bam -a GRCh37_to_GRCh38.chain.gz Test5.hg19.bam output.hg38
Add tags: True
Insert size = 200.000000
Insert size stdev = 30.000000
Number of stdev from the mean = 3.000000
Add tags to each alignment = True
2024-01-12 09:29:11 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
[E::idx_find_and_load] Could not retrieve index file for 'Test5.hg19.bam'
2024-01-12 09:29:11 [INFO] Liftover BAM file "Test5.hg19.bam" to "output.hg38.bam"
2024-01-12 09:29:15 [INFO] Done!
2024-01-12 09:29:15 [INFO] Sort "output.hg38.bam" and save as "output.hg38.sorted.bam"
2024-01-12 09:29:15 [INFO] Index "output.hg38.sorted.bam" ...
Total alignments: 99,914
QC failed: 0
Paired-end reads:
R1 unique, R2 unique (UU): 96035
R1 unique, R2 unmapped (UN): 3638
R1 unique, R2 multiple (UM): 0
R1 multiple, R2 multiple (MM): 0
R1 multiple, R2 unique (MU): 230
R1 multiple, R2 unmapped (MN): 11
R1 unmapped, R2 unmapped (NN): 0
R1 unmapped, R2 unique (NU): 0
R1 unmapped, R2 multiple (NM): 0
Tag Definitions
General Tags
Tag |
Meaning |
|---|---|
Q |
QC failed. |
N |
Unmapped (either originally unmapped or failed liftover). |
M |
Multiple mapped (can be lifted over to multiple locations). |
U |
Unique mapped (lifted over to a single location). |
Paired-End Tags
QF — QC failed
NN — both reads unmapped
NU — read1 unmapped, read2 uniquely mapped
NM — read1 unmapped, read2 multiply mapped
UN — read1 uniquely mapped, read2 unmapped
UU — both reads uniquely mapped
UM — read1 uniquely mapped, read2 multiply mapped
MN — read1 multiply mapped, read2 unmapped
MU — read1 multiply mapped, read2 uniquely mapped
MM — both reads multiply mapped
Single-End Tags
QF — QC failed
SN — unmapped
SM — multiply mapped
SU — uniquely mapped
Note
All alignments (mapped, partially mapped, unmapped, or QC failed) are written to a single output file. Users can filter alignments using the assigned tags.
The header section is updated to reflect the target assembly, CrossMap version, and chain file used.
Genome coordinates and all SAM flags in the alignment section are updated accordingly.
When using CRAM input, ensure
pysam >= 0.8.2.Optional fields in the alignment section are not modified.
Convert Wiggle Format Files
The Wiggle (WIG) format is used to display continuous, quantitative data along the genome — for example, GC content or read coverage from high-throughput sequencing experiments.
BigWig is the binary, indexed version of Wiggle, providing faster random access and efficient storage.
CrossMap supports both variableStep (irregular intervals) and fixedStep (regular intervals) Wiggle files as input. Regardless of the input format, the output is always a bedGraph file.
Command Help
To display the help message for Wiggle file conversion, run CrossMap wig -h
usage: CrossMap wig [-h] [--chromid {a,s,l}] input.chain input.wig out_wig
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
Can be a plain text file or compressed
(.gz, .Z, .z, .bz, .bz2, .bzip2).
input.wig Input Wiggle or bedGraph file
(http://genome.ucsc.edu/goldenPath/help/wiggle.html).
Supports both “variableStep” and “fixedStep” formats.
May be plain text, compressed (.gz, .bz2, etc.),
or a URL (http://, https://, ftp://).
*Compressed remote files are not supported.*
out_wig Output bedGraph file.
Regardless of input (Wiggle or bedGraph),
the output is always in bedGraph format.
options:
-h, --help Show this help message and exit
--chromid {a,s,l} Style of chromosome IDs:
a = as-is
l = long style (e.g., "chr1", "chrX")
s = short style (e.g., "1", "X")
Note
To improve performance, CrossMap internally calls the GNU sort command. If the system “sort” command is unavailable or not callable, CrossMap will terminate with an error.
Convert BigWig Format Files
The BigWig format is a binary, indexed version of the Wiggle (WIG) format that allows for efficient random access and visualization of continuous genomic data such as read depth or coverage.
CrossMap can lift over BigWig files between assemblies using a chain file.
Command Help
To display the help message for BigWig file conversion, run CrossMap bigwig -h
usage: CrossMap bigwig [-h] [--chromid {a,s,l}] input.chain input.bw output.bw
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
Can be a plain text file or compressed
(.gz, .Z, .z, .bz, .bz2, .bzip2).
input.bw Input BigWig file
(https://genome.ucsc.edu/goldenPath/help/bigWig.html).
output.bw Output BigWig file.
options:
-h, --help Show this help message and exit
--chromid {a,s,l} Style of chromosome IDs:
a = as-is
l = long style (e.g., "chr1", "chrX")
s = short style (e.g., "1", "X")
Example
$ CrossMap bigwig GRCh37_to_GRCh38.chain.gz Test6.hg19.bw output.hg38
2024-01-12 09:37:32 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
2024-01-12 09:37:33 [INFO] Liftover BigWig file "Test6.hg19.bw" to bedGraph file "output.hg38.bgr"
2024-01-12 09:37:33 [INFO] Merging overlapped entries in bedGraph file
2024-01-12 09:37:33 [INFO] Sorting bedGraph file: output.hg38.bgr
2024-01-12 09:37:33 [INFO] Writing header to "output.hg38.bw" ...
2024-01-12 09:37:33 [INFO] Writing entries to "output.hg38.bw" ...
Note
CrossMap internally calls the GNU sort command to improve performance during sorting and merging. If the system “sort” command is not available, CrossMap will terminate with an error.
Convert GFF / GTF Format Files
The GFF (General Feature Format) and GTF (Gene Transfer Format) are text-based formats used to describe genomic features such as genes, exons, introns, and regulatory elements.
GTF is a refined version of GFF, and both share the same first eight fields.
CrossMap supports plain text, compressed files, and URLs pointing to remote
resources as input (http://, https://, or ftp://).
Note: compressed remote files are not supported.
During liftover, CrossMap updates only the chromosome names and genome coordinates. The output format (GFF or GTF) matches the format of the input file.
Command Help
To display the help message for GFF/GTF conversion, run CrossMap gff -h
usage: CrossMap gff [-h] [--chromid {a,s,l}] input.chain input.gff [out_gff]
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
Can be plain text or compressed (.gz, .Z, .z, .bz, .bz2, .bzip2).
input.gff Input GFF (General Feature Format) or GTF (Gene Transfer Format) file.
Supports plain text, compressed (.gz, .bz2, etc.), or URLs
(http://, https://, ftp://). Compressed remote files are not supported.
out_gff Output GFF/GTF file. If omitted, CrossMap writes the result to STDOUT.
options:
-h, --help Show this help message and exit
--chromid {a,s,l} Style of chromosome IDs:
a = as-is
l = long style (e.g., "chr1", "chrX")
s = short style (e.g., "1", "X")
Example
$ CrossMap gff GRCh37_to_GRCh38.chain.gz Test7.hg19.gtf output.hg38
2024-01-12 09:43:53 [INFO] Read the chain file "GRCh37_to_GRCh38.chain.gz"
Note
Each genomic feature (exon, intron, UTR, etc.) is processed independently. CrossMap does not verify whether features that originally belonged to the same gene still map to the same gene after conversion.
To lift over complete gene annotation files, use the BED12 format instead.
If no output file is specified, the converted results are printed to the console. In this case, entries that fail to convert are also displayed.
Convert VCF Format Files
The VCF (Variant Call Format) is a standardized, line-oriented text format developed by the 1000 Genomes Project. It is widely used for representing single nucleotide variants (SNVs), insertions/deletions (indels), copy number variations (CNVs), and structural variants.
CrossMap updates chromosome names, genome coordinates, and reference alleles to match a new assembly. All other fields in the VCF record remain unchanged.
Command Help
To display the help message for VCF conversion:
CrossMap vcf -h
Example output:
usage: CrossMap vcf [-h] [--chromid {a,s,l}] [--no-comp-alleles] [--compress]
input.chain input.vcf refgenome.fa out_vcf
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
Can be plain text or compressed
(.gz, .Z, .z, .bz, .bz2, .bzip2).
input.vcf Input VCF file
(https://samtools.github.io/hts-specs/VCFv4.2.pdf).
Supports plain text, compressed (.gz, .bz2, etc.), or URLs
(http://, https://, ftp://). *Compressed remote files are not supported.*
refgenome.fa Reference genome sequence of the **target assembly**
in FASTA format.
out_vcf Output VCF file.
options:
-h, --help Show this help message and exit
--chromid {a,s,l} Style of chromosome IDs:
a = as-is
l = long style (e.g., "chr1", "chrX")
s = short style (e.g., "1", "X")
--no-comp-alleles Skip checking whether the reference allele differs
from the alternate allele.
--compress Compress the output VCF file using the system "gzip" utility.
Notes
Note
Genome coordinates and reference alleles are updated to match the target assembly.
The reference genome file provided must correspond to the target assembly.
If the reference FASTA file (e.g.,
../database/genome/hg18.fa) is not indexed, CrossMap will automatically index it during the first run.- CrossMap produces two output files:
output_file— the converted VCFoutput_file.unmap— variants that could not be mapped
Chromosome ID style in the output (e.g.,
chr1vs.1) matches the input format.
Convert MAF Format Files
The MAF (Mutation Annotation Format) is a tab-delimited text format used to describe somatic and/or germline mutation annotations. Please do not confuse it with the Multiple Alignment Format (MAF), which is unrelated.
CrossMap supports MAF files in plain text, compressed form, or accessible via URLs
(http://, https://, ftp://).
Note: compressed remote files are not supported.
Command Help
To display the help message for MAF file conversion, run CrossMap maf -h
usage: CrossMap maf [-h] [--chromid {a,s,l}]
input.chain input.maf refgenome.fa build_name out_maf
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
May be plain text or compressed
(.gz, .Z, .z, .bz, .bz2, .bzip2).
input.maf Input MAF file
(https://docs.gdc.cancer.gov/Data/File_Formats/MAF_Format/).
Can be plain text, compressed (.gz, .bz2, etc.), or a URL
(http://, https://, ftp://). *Compressed remote files are not supported.*
refgenome.fa Reference genome sequences of the **target assembly**
in FASTA format.
build_name Name of the **target assembly** (e.g., "GRCh38").
out_maf Output MAF file.
options:
-h, --help Show this help message and exit
--chromid {a,s,l} Style of chromosome IDs:
a = as-is
l = long style (e.g., "chr1", "chrX")
s = short style (e.g., "1", "X")
Note
CrossMap updates chromosome names, coordinates, and reference alleles in the MAF file to match the target assembly.
The reference genome FASTA file must correspond to the target build name provided.
- Output files:
output_file— converted MAF fileoutput_file.unmap— entries that failed to map
Convert GVCF Format Files
A GVCF (Genomic Variant Call Format) is an extension of the standard VCF format used by tools such as GATK to store both variant and non-variant regions.
CrossMap can lift over GVCF files by updating chromosome names, coordinates, and reference alleles while keeping all other fields unchanged.
Command Help
To display the help message for GVCF conversion, run CrossMap gvcf -h
usage: CrossMap gvcf [-h] [--chromid {a,s,l}] [--no-comp-alleles] [--compress]
input.chain input.gvcf refgenome.fa out_gvcf
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
May be plain text or compressed
(.gz, .Z, .z, .bz, .bz2, .bzip2).
input.gvcf Input GVCF file
(https://samtools.github.io/hts-specs/VCFv4.2.pdf).
Supports plain text, compressed (.gz, .bz2, etc.), or URLs
(http://, https://, ftp://). *Compressed remote files are not supported.*
refgenome.fa Reference genome sequences of the **target assembly**
in FASTA format.
out_gvcf Output GVCF file.
options:
-h, --help Show this help message and exit
--chromid {a,s,l} Style of chromosome IDs:
a = as-is
l = long style (e.g., "chr1", "chrX")
s = short style (e.g., "1", "X")
--no-comp-alleles Skip checking whether the reference allele differs
from the alternate allele.
--compress Compress the output GVCF file using the system "gzip".
Note
Genome coordinates and reference alleles are updated to the target assembly.
The reference FASTA file must correspond to the target assembly.
If the reference genome is not indexed, CrossMap will automatically index it.
- Output files:
output_file— converted GVCF fileoutput_file.unmap— entries that failed to map
The chromosome ID format in the output (e.g.,
chr1vs.1) matches the input format.
Convert Large Genomic Regions
For large genomic regions (such as CNV blocks), the default
CrossMap bed command splits each large region into smaller segments
that can be 100% mapped to the target assembly.
In contrast, CrossMap region does not split regions.
Instead, it calculates the mapping ratio — defined as:
If the computed map ratio exceeds the threshold specified by the
-r (or --ratio) option, the region is considered successfully mapped
and its coordinates are converted to the target assembly.
Otherwise, the conversion fails and the region is recorded as unmapped.
Command Help
To view the help message for converting large genomic regions, run CrossMap region -h
usage: CrossMap region [-h] [--chromid {a,s,l}] [-r MIN_MAP_RATIO]
input.chain input.bed [out_bed]
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
Can be a plain text file or compressed
(.gz, .Z, .z, .bz, .bz2, .bzip2).
input.bed Input BED file.
The first three columns must be “chrom”, “start”, and “end”.
Supports plain text, compressed (.gz, .bz2, etc.), or URLs
(http://, https://, ftp://). *Compressed remote files are not supported.*
out_bed Output BED file.
If omitted, CrossMap writes results to STDOUT.
options:
-h, --help Show this help message and exit
--chromid {a,s,l} Style of chromosome IDs:
a = as-is
l = long style (e.g., "chr1", "chrX")
s = short style (e.g., "1", "X")
-r, --ratio MIN_MAP_RATIO
Minimum ratio of bases that must successfully remap.
Regions with a lower ratio will be reported as unmapped.
Notes
Note
The default minimum mapping ratio (
-r) is 0.85 (i.e., 85% of bases must map).This command is ideal for converting large genomic blocks such as copy number variation (CNV) regions, segmental duplications, or long structural variants.
If an output file is not specified, results are printed to the console.
Unmapped or partially mapped regions are written to the
*.unmapfile.
View Chain File
The CrossMap viewchain command converts a UCSC
chain file
into a simple, tab-delimited format that is easier to read and interpret.
Each line in the output represents a block-to-block alignment
between two genome assemblies.
This function is useful for inspecting or debugging chain files, visualizing how source and target regions align, or verifying liftover results.
Command Help
To display the help message for the viewchain command:
CrossMap viewchain -h
Example output:
usage: CrossMap viewchain [-h] input.chain
positional arguments:
input.chain Chain file
(https://genome.ucsc.edu/goldenPath/help/chain.html)
Describes pairwise alignments between two genomes.
May be a plain text file or compressed
(.gz, .Z, .z, .bz, .bz2, .bzip2).
options:
-h, --help Show this help message and exit
Example
$ CrossMap viewchain GRCh37_to_GRCh38.chain.gz > chain.tab
$ head chain.tab
1 10000 177417 + 1 10000 177417 +
1 227417 267719 + 1 257666 297968 +
1 317719 471368 + 1 347968 501617 -
1 521368 1566075 + 1 585988 1630695 +
1 1566075 1569784 + 1 1630696 1634405 +
1 1569784 1570918 + 1 1634408 1635542 +
1 1570918 1570922 + 1 1635546 1635550 +
1 1570922 1574299 + 1 1635560 1638937 +
1 1574299 1583669 + 1 1638938 1648308 +
1 1583669 1583878 + 1 1648309 1648518 +
Note
Each line represents an alignment block between the source and target assemblies.
- Columns correspond to:
Source chromosome, start, end, and strand
Target chromosome, start, end, and strand
The output file is plain text and can be easily parsed for further analysis.
Comparison to UCSC LiftOver Tool
To evaluate the accuracy of CrossMap, we randomly generated 10,000 genomic intervals (each 200 bp in length) from the hg19 assembly. The dataset can be downloaded from here.
We then converted these intervals to hg18 coordinates using both CrossMap and the UCSC LiftOver tool, under default configuration settings.
The UCSC LiftOver tool was chosen for comparison because it is the most widely used method for genome coordinate conversion.
Results Summary
CrossMap failed to convert: 613 intervals
UCSC LiftOver failed to convert: 614 intervals
All failed intervals were identical between the two tools, except for one region (
chr2:90542908–90543108).
For that single region, UCSC failed because the interval needed to be split twice during liftover, while CrossMap successfully handled the multi-split mapping.
Original (hg19) |
Split (hg19) |
Target (hg18) |
|---|---|---|
chr2 90542908 90543108 - |
chr2 90542908 90542933 - |
chr2 89906445 89906470 - |
chr2 90542908 90543108 - |
chr2 90542933 90543001 - |
chr2 87414583 87414651 - |
chr2 90542908 90543108 - |
chr2 90543010 90543108 - |
chr2 87414276 87414374 - |
For all other successfully converted regions, the start and end coordinates were identical between CrossMap and UCSC LiftOver.
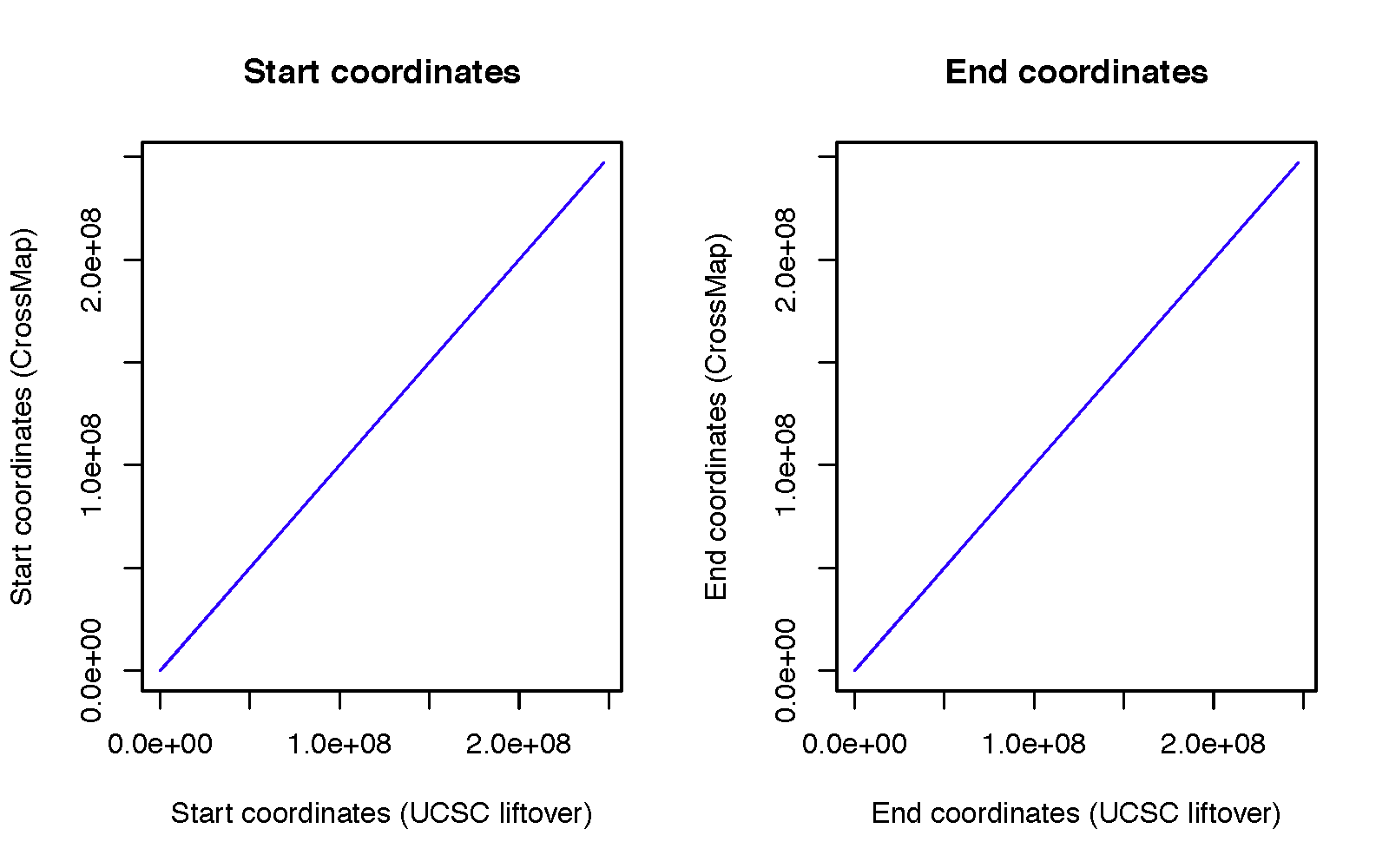
Figure: Comparison of CrossMap and UCSC LiftOver conversions between hg19 and hg18 assemblies.
Citation
If you use CrossMap in your research or publications, please cite:
Zhao, H., Sun, Z., Wang, J., Huang, H., Kocher, J.-P., & Wang, L. (2013). CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics (Oxford, England), btt730. https://pubmed.ncbi.nlm.nih.gov/24351709/
LICENSE
CrossMap is distributed under the GNU General Public License (GPL).
This program is free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation — either version 2 of the License, or (at your option) any later version.
CrossMap is distributed in the hope that it will be useful, but without any warranty — without even the implied warranty of merchantability or fitness for a particular purpose.
See the GNU General Public License for more details.
You should have received a copy of the GNU General Public License along with this program. If not, write to:
Free Software Foundation, Inc.
51 Franklin Street, Fifth Floor
Boston, MA 02110-1301 USA